Subcellular Fractionation
Eukaryotic cells are composed of a variety of organelles having different functions. In a biochemical examination of organelles, purified samples are required hence the need for isolation. The most common isolation technique is differential centrifugation. During isolation, the cells are homogenized so as to break open the cell membrane. Homogenization can be achieved by crushing, filtration, grinding, sonication, and solubilization (Diels and Chris 203). The method used for homogenization is determined by the tissue type and objective of the experiment. In homogenization, isotonic sucrose is used to avoid rupturing the membranes by osmotic pressure (Wilson and Walker 105). In this practical, density gradient centrifugation technique was used to isolate crude subcellular fractions from rabbit liver. Under density gradient centrifugation, the cells are ruptured mechanically, followed by centrifugation at low speeds to remove cellular debris and large organelle components. Differential centrifugation at higher speeds isolates a crude mitochondrial extract for enzymatic analysis.
Aim
The purpose of the experiment was to isolate mitochondria from rabbit liver using the density gradient centrifugation technique.
Materials
The materials used in this experiment were a mito-buffer, ice bucket, micro-centrifuge, sterile 2ML microfuge tubes, tip discard, filter tips, pipettes, and Vortex mixers.
Pre-lab Preparation by Technical Staff
The rabbit liver was pre-prepared by the technical staff prior to the experiment. This preparation was performed to maintain their integrity and function. The preparation procedures included chopping the liver into small pieces and then placing it in sucrose solution. The tissue was then homogenized and stored in liquid nitrogen.
Student Procedure
Sterile screw cap tubes with lids were prepared and labeled as WCL (whole cell lysate), SN1 (supernatant 1), SN2 (supernatant 2), SN3 (supernatant 3), and Mito (mitochondrial fraction). The tubes were stored on ice until required so that they were pre-chilled. The cell lysate was transferred to the tube labeled WCL and stored on ice.
The cell debris in the homogenate (1.9 mL) was pelleted at 1000×g for 10 minutes. The supernatant was transferred to the labeled Mito and centrifuged at 10 min at 10000 ×g. The supernatant was then transferred to the tube labeled SN1 and kept on ice. The pellet was suspended, and then 1000 µL of the mito-buffer was added and centrifuged at 12000 ×g for 10 minutes. The supernatant was transferred using a pipette into the tube labeled SN2 and stored on ice. The pellet was re-suspended well, then 400 µL of the mito-buffer added to the mitochondrial pellet, mixed well, and stored on ice. For protein assay, 10 µL of WCL, SN1, SN2, SN3, and Mito samples were taken then the fractions were transferred immediately to 80 °C for the next practical session.
BCA Protein Assay
Aim
This essay aimed to determine the concentration of protein samples by use of a standard curve.
Materials
The materials required included bovine serum albumin, spectrophotometer, protein samples, microtitre plate, and buffered saline.
Method
Bovine serum albumin standards were used to prepare protein standards. From each standard, 25 µL was transferred into respective wells, then 15 µL of Mito buffer was added into the wells. A color reagent was placed into each well. The plate was incubated for 30 minutes at 37° C before measuring the absorbance at 595 nm. The data obtained was used to draw the standard curve below.
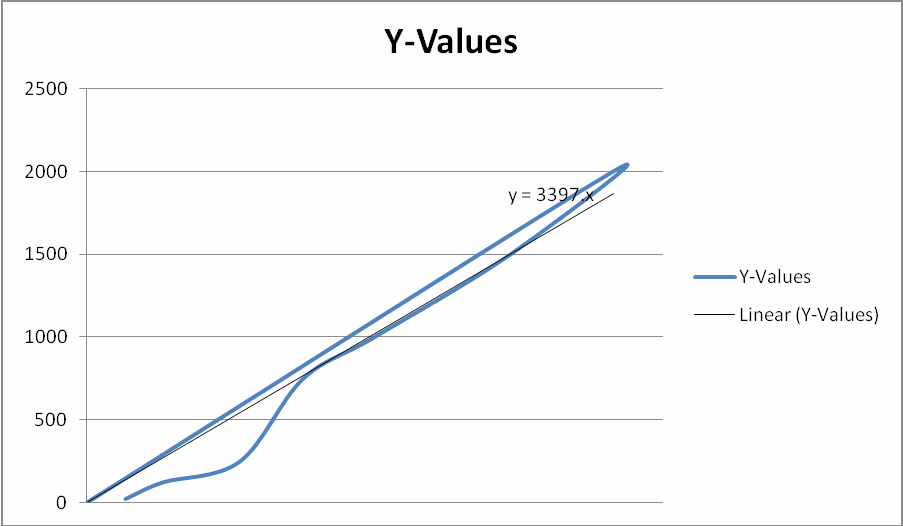
Data Analysis: Estimating Protein Concentration in the Sample
The standard curve was used in estimating the amount and concentration of the protein samples by comparing their absorbance with those of the standard protein. The data obtained were tabulated as shown below.
Table: Concentration of Protein Samples.
Discussion
Subcellular fractionation techniques are essential in elucidating the functions of the various components of the cell as it facilitates biochemical studies to be performed. The plasma membranes are disrupted without affecting the internal constituents of the cell. On centrifugation, pellets are formed depending on size and density, resulting to the recovery of concentrated fractions (Wilson and Walker 118). In spite of the strengths of subcellular fractionation, the technique cannot achieve complete purification. This is because some of the fractions share physical properties and hence can fractionate almost at the same gradients. The technique cannot also produce free intact organelles thus contamination with other organelles is often observed (Graham and Rickwood 39).
Succinate Dehydrogenase Assay
The mitochondrion plays a role in cellular respiration through its biochemical machinery. One of the biochemical machinery in the mitochondria is Succinate dehydrogenase enzyme, an enzyme of citric acid cycle which converts succinate to fumarate. Succinate dehydrogenase tightly binds the inner mitochondrial membrane, together with flavin adenine dinucleotide which is its coenzyme. This enzyme is present in the mitochondrion of eukaryotes in stable and large amounts hence can be used as a marker enzyme. This can be done by assaying the enzyme’s activity using an artificial electron acceptor to show color changes (Wilson and Walker 123). The artificial electron acceptor is reduced by this enzyme to formazan, which is photometrically determined at 490nm. The enzyme’s activity is visible with naked eyes since the solution changes to be rusty red.
Aim
The aim of this assay was to demonstrate the presence or absence of mitochondria in isolated sub cellular fractions by assaying each fraction for the enzyme Succinate dehydrogenase.
Materials
The materials that were used included; sub-cellular fractions, succinate solution, electron acceptor, stop solution, glass cuvettes, and spectrophotometer.
Procedure
300 µL of succinate solution was put into 10 sterile, labeled tubes and 50 µG of each sub-cellular fraction added to the respective tubes. The samples and blanks were incubated at 37 °c for 10 minutes then a stop solution added to the blank tubes. An artificial electron acceptor (p-iodonitrotetrazolium) was added into the tubes, followed by incubation for half an hour at 37°C. A stop solution was added into the tubes labeled assay which was then centrifuged and the supernatant transferred to glass cuvetted. The absorbance of each sample at 490 nm was determined against the corresponding blank.
Table: Absorbance of Assay and Blank.
Sodium Dodecyl Sulfate (SDS) PolyAcrylamide Gel Electrophoresis
PolyAcrylamide gel electrophoresis is an analytical technique for purification of proteins from a mixture based on their size and electric charge. A charged molecule moves in an electric field to the pole with opposite charge. Protein mobility is dependent on, among other factors, whether the protein is positively or negatively charged and the size of the molecule. To determine the molecular weight of the proteins, the samples are treated with an anionic detergent called Sodium Dodecyl Sulfate (SDS) to impart them with same charge. SDS denatures the molecules so that their electrophoretic mobility is determined by size, hence enabling estimation of molecular weight (Simpson 43). SDS gives the proteins a negative charge by binding to them without altering the peptide binds. To denature the proteins, the mixture is heated in 2-mercaptoethanol buffer. SDS as well as a reducing agent is also components of the buffer. The role of the mercaptoethanol is beaks the cysteine disulfide bonds while bonds between proteins are disrupted by the SDS. This results into production of individual polypeptide chains, which once placed in a supporting medium like polyacrylamide in an electric field, the protein moves towards the positively charged terminal. Through molecular sieving effect, the proteins separate depending on their size. The separated molecules are stained with protein specific technique to enhance visualization then the size determined by examining the migration distance with a standard marker (Simpson 47).
Materials
The materials that were required for this activity included acrylamide gel, gel loading buffer, running buffer, protein samples, staining solutions, power pack, pipettor.
Procedure
20 µg of each of the fractions was mixed with some amount of SDS PAGE sample buffer in a labeled microfuge. The samples were centrifuged gently and rested before loading them onto the gel. The electrophoresis apparatus was set and run at 200 Volts. The gel was stuck into the plates then some running buffer added. The gel was stained and destained before being scanned.
References
Diels, Ann and Chris Michiels. “High-pressure homogenization as a non-thermal technique for the inactivation of microorganisms.” Critical Reviews in Microbiology 32.4 (2006): 201-216. MEDLINE. Web.
Graham, John and Rickwood David. Subcellular fractionation: a practical approach. New York, NY: Oxford University Press, 1997. Print.
Simpson, Richard. Proteins and proteomics: a laboratory manual. Woodbury, NY: CSHL Press, 2003. Print.
Wilson, Keith and Walker John. Principles and techniques of biochemistry and molecular biology, 7th Ed. New York, NY: Cambridge University Press, 2010. Print.
