One of the most critical tasks of modern bioinformatics is to predict and foretell the structure and, consequently, the functions of bacterial proteins. To reveal amino acid followers, -helix and -sheet location, as a rule, computer programming methods are used. Protein prediction is an incredibly important goal since it allows to simplify biotechnological research in the process of designing new drugs or enzymes. On the other hand, many strains of bacteria are pathogenic and can significantly harm human and animal health. For this reason, specialists are interested in anticipating a protein abnormal to the host organism in the carrier bacterium so that a drug can be developed in time. In addition, knowledge of protein structure may prompt potential partners for protein interaction and thus encourage researchers to develop or improve new enzymes or antibodies, or, for example, to explain the phenotype of the mutations performed or to help determine the location of the mutations in order to change specific phenotypes.
Traditionally, prediction of the functions of bacterial proteins is carried out for poorly studied molecules or hypothetical proteins predicted based on these genome sequences. The source of information for the prediction can be the homology of nucleotide sequences, gene expression profiles or phylogenetic and phenotypic profiles. Taking into account multi-variant protein functions, the task of accurate and analytical prediction is one of the most important directions of modern bioinformatics.
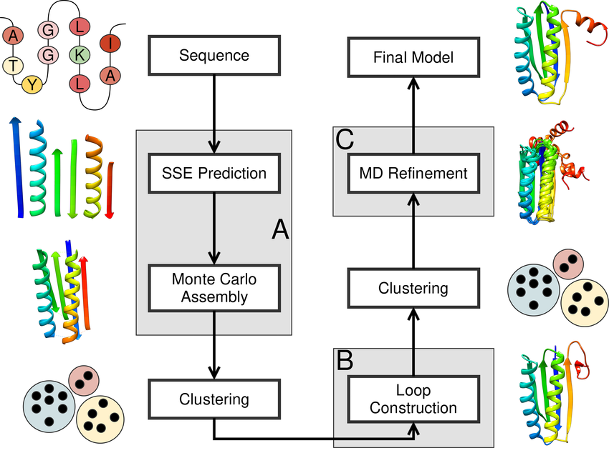
Prediction of protein structure and functions is a complex task for two reasons. First, the number of possible spatial configurations of proteins is incredibly vast, and secondly, the physical foundations of protein structuring and its stability have not been thoroughly investigated. Nevertheless, some workings out are already available: if among known structures it is possible to find such, for which there are bases to assert, that they can be in a certain degree similar to the object of modelling, then they can be used as a template for construction. This type of method is called template-based modelling: templates can be found through comparison of amino acid sequences in software BLAST (in particular, BlastP), using text format FASTA. It is necessary to carry out pairwise alignment, which allows revealing conservative residues in the whole family or separate subfamilies of proteins.
Modelling by template has a vast practical potential because if the structure of at least one protein from which functional family is known, then it is possible to try to build models for almost every protein in this family. With the growth of the structure database, such modelling becomes possible for more and more proteins. On the other hand, if it is not possible to find a template for modelling protein structure, then physical and chemical methods are used. However, such methods do not have high reliability; hence more often, specialists turn to the first method.
